
Page 117 - 201905
P. 117
第 5 期 刘 军,等: 基于 DFT 和 UV-Vis 的金丝桃苷印迹相互作用分析 ·885·
氢原子与 HYP 上酮基氧原子,IA 上羰基氧原子与 从表 1 中可以看出,不同印迹比例下,分子间
HYP 上羟基氢原子可能发生相互作用。正是由于它 的氢键范围为 0.1631~0.2081 nm,所有氢键均为强
们直接相反的电子性质可能导致 HYP 与 AM(或 氢键,有利于印迹效率的提高;但所形成的氢键键
HYP 与 IA)之间强烈的静电吸引力,这也是构建分 长发生了一些细微的变化,这种差异主要是由于分
子印迹聚合物可能的最初原始动力。结合 HYP、AM 子构象的稳定化而导致所形成的复合物中各原子的
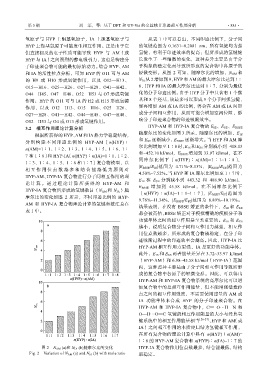
和 IA 的活性位点分析,可知 HYP 的 O11 可与 AM 轻微变形。从图 2 可知,随摩尔比的增加,N AM 和
的 H9 或 H10 形成氢键作用,以及 O12—H13、 N IA 从 2 增加到 9,HYP 和 AM 的最大摩尔比达到 1∶
O15—H16、O25—H26、O27—H28、O41—H42、 8,HYP 和 IA 的最大摩尔比达到 1∶7,分别为最优
O44—H45、O47—H48、O52—H53 与 O7 形成氢键 化的分子印迹比例。由于 HYP 分子中只含有 1 个羰
作用;HYP 的 O11 可与 IA 的 H2 或 H15 形成氢键 基和 8 个羟基,故最多可以形成 9 个分子间强氢键,
作用,以及 O12—H13、O15—H16、O25—H26、 如再增加 AM 或 IA 的比例,将会在 AM 或 IA 间形
O27—H28、O41—H42、O44—H45、O47—H48、 成分子间相互作用,从而可能会增加空间位阻,影
响分子印迹聚合物的印迹吸附效率。
O52—H53 与 O4 或 O13 形成氢键作用。
3.2 相互作用理论计算分析 HYP-AM 和 HYP-IA 复合物的 E IE 、E SE 、E BSSE
根据所获得的 HYP、AM 和 IA 热力学稳定结构, 随摩尔比的变化如图 3 所示,随摩尔比的增加,E IE
和 E SE 逐渐减小,E BSSE 逐渐增大。当 HYP 和 AM 摩
分别构建不同印迹比例的 HYP-AM〔 n(HYP)∶
尔比例增加至 1∶8 时,E IE 和 E SE 分别减小到418.13
n(AM)=1∶1、1∶2、1∶3、1∶4、1∶5、1∶6、1∶
和452.10 kJ/mol,E BSSE 增加到 33.97 kJ/mol,在不
7 和 1∶8〕和 HYP-IA〔n(HYP)∶n(IA)=1∶1、1∶2、
同摩尔比例下〔n(HYP)∶ n(AM)=1∶ 1~1∶8〕,
1∶3、1∶4、1∶5、1∶6 和 1∶7〕复合物模型,以
|E BSSE /E IE |范围为 4.71%~8.13%,|E BSSE /E SE |范围为
相 互作用 位点 数越多 和结 合能越 低为 原则 对
4.50%~7.52%。当 HYP 和 IA 摩尔比增加至 1∶7 时,
HYP-AM、HYP-IA 复合物进行分子间相互作用的理
E IE 和 E SE 分别减小到443.32 和488.90 kJ/mol,
论计算。通 过理论计算 所获得的 HYP-AM 和
E BSSE 增加到 45.58 kJ/mol,在不同 摩尔比例下
HYP-IA 复合物所形成的氢键数目(N AM 和 N IA )随
〔n(HYP)∶n(IA)=1∶1~1∶7〕,|E BSSE /E IE |范围为
摩尔比的变化如图 2 所示,不同印迹比例的 HYP-
9.76%~11.34%,|E BSSE /E SE |范围为 8.89%~10.19%。
AM 和 HYP-IA 复合物理论计算的氢键参数汇总在
结果表明,在没有 BSSE 矫正的条件下,E IE 和 E SE
表 1 中。 都会被高估,BSSE 矫正对于模拟精确的模板分子和
功能单体之间的相互作用是至关重要的。E IE 和 E SE
越小,说明复合物分子间相互作用力越强,相互作
用位点数越多,所形成的复合物越稳定,在分子印
迹吸附过程中的印迹效率会越高。因此,HYP-IA 比
HYP-AM 相互作用力更强,IA 是更好的功能单体。
此外,E IE 和 E SE 两者能量差异在 3.72~33.97 kJ/mol
(HYP-AM)和 6.98~45.58 kJ/mol(HYP-IA)范围
内,这种差异主要是由于分子间相互作用导致所形
成的复合物中各原子的轻微变形。因此,可以得出
HYP-AM 和 HYP-IA 复合物系统的高摩尔比可以增
加复合物中的总相互作用能量,但不能增强活性位
点之间的相互作用强度,不需要使用过量的 AM 或
IA 功能单体来合成 HYP 的分子印迹聚合物。在
HYP-AM 和 HYP-IA 复合物中,C== O···H—N 和
O—H···O==C 氢键的相互作用能量的大小与经典氢
键系统中的相互作用能量相当 [26-27] ,HYP 和 AM(或
IA)之间相互作用的本质应归结为氢键相互作用,
在所有复合物的理论计算中具有 n(HYP)∶n(AM)=
1∶8 的 HYP-AM 复合物和 n(HYP)∶n(IA)=1∶7 的
图 2 N AM (a)和 N IA (b)随摩尔比的变化 HYP-IA 复合物作用位点数最多,结合能最低,结构
Fig. 2 Variation of N AM (a) and N IA (b) with mole ratio 最稳定。